

【罕見病/01】罕見病群體不因微小而沉默




罕見病,雖然顧名思義是指患者人數極少的疾病,可是目前世界已知的罕見病有七千多種,換句話說,如果把所有種類的罕見病患者加起來,那會是一個不小的群體。
ADVERTISEMENT
可悲的是,約30%罕見病患者未到5歲就已過世,即使得以存活,患者往往也會面臨漫長而驚險的求醫之路,有些罕見病甚至動輒需要幾十萬乃至幾百萬令吉醫藥費。
患者少,不意味罕見病就微不足道。如果說“勿因善小而不為”,那麼每一個渺小的罕見病患者,也不應該因為罕見而被我們視而不見。
報道:本刊 梁慧穎
攝影:本報 黃冰冰
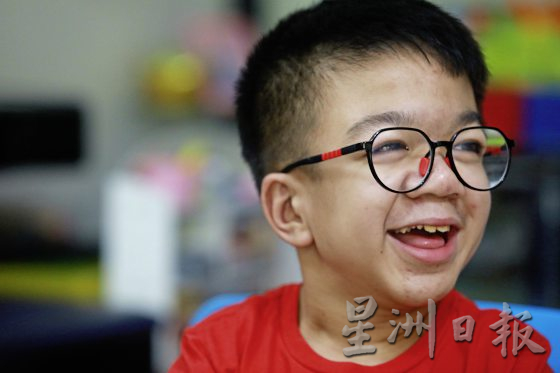
2015年,當時只有3歲的張嘉健被診斷患有第一型黏多糖症(MPS type I,或Hurler syndrome),急需60萬令吉進行非親屬骨髓移植手術,若不盡快動刀,他很有可能活不過9歲。
第一型黏多糖症是一種罕見病,在嘉健3歲以前,他身體其實已經出了一些狀況,才9個月大就動過兩次小腸氣手術和一次腦部手術。然而,即使他去過很多醫院看過很多醫生,都沒有一位醫生能夠明確說出他到底得了什麼病。直到他去馬大醫藥中心經過臨床遺傳學家的診斷,才證實患有罕見病。

當時他有兩種治療方案:一種是酶替代療法(ERT),估計每星期要去醫院打藥一次;另一種則是一次性的骨髓移植手術。父親張志勝說,酶替代療法的醫藥費非常昂貴,以嘉健當時3歲的體重和身軀,每年醫藥費就高達30萬至50萬令吉。當時就診的吉隆坡中央醫院說“要等”,但要等到何年何月院方也不確定,所以他和太太決定讓孩子接受風險不低的骨髓移植手術。
為了手術,他們一家登上新聞版面,懇請社會大眾慷慨解囊。幸好那次手術相當成功,可是畢竟嘉健的眼睛和心臟在動手術前就已受損,所以這些病症會一直駐留他弱小的身體裡面,未來甚至有可能會惡化。
儘管前方道路依然崎嶇,但他父母聲聲感恩,因為至少他現在每天還能開開心心到特殊班上課,比起其他早逝或無法走動的罕見病小孩,他們知道嘉健還不是最不幸的一位。



誰可能會得罕見病?
目前世界已知的罕見病超過7000種,不同國家對罕見病的定義有所不同,在我國,如果根據馬來西亞罕見病協會(MRDS)的定義,每4000人中有1人得病即屬於罕見病。
很多罕見病在患者幼年時發病,約30%患者會在5歲之前病逝,即使得以存活,往往也需要長期的醫療照護。馬大醫藥中心臨床遺傳學家湯茂強教授強調:“罕見病可以發生在任何人身上,無論成人或小孩;無論窮人或富人;無論男性或女性;也不管是什麼種族。這個觀念很重要,因為有些人以為罕見病只會影響嬰兒,其實是不正確的。”

根據研究,約80%罕見病屬於遺傳病,其餘20%是環境汙染等其他因素造成。湯茂強舉例,好萊塢電影《惡水真相》(Minamata)描述水銀汙染的真實事件,造成上世紀日本熊本縣水俁市許多嬰兒一出生就得了罕見病。另外,孕婦懷孕期間若感染寨卡病毒,也有可能導致胎兒出現小頭症,總之罕見病未必是遺傳造成。
哪些罕見病比較常見?
在我國,比較常見的罕見病有以下5種:
●馬凡氏症候群(Marfan syndrome)
●普瑞德威利症候群(Prader-Willi syndrome,俗稱小胖威利)
●成骨不全症(Osteogenesis imperfecta,俗稱玻璃娃娃)
●MELAS症候群
●第二型黏多糖症(MPS type II,Hunter syndrome)
(資料來源:www.rarediseasemalaysia.com)
然而,湯茂強不想太強調哪種病屬於5大罕見病,因為他相信還有一些罕見病患者沒有到醫院求醫,所以社會根本不知道這些人的存在。如果我們過於強調5大罕見病,他擔心其他罕見病群體也許會遭到擠壓或被忽視。

延伸閱讀: 【罕見病/02】罕見病病患的困境 【罕見病/03】減少罕見病病例 及早預防勝於治療 相關稿件: 【醫院營養餐/01】醫院“千人宴” 營養優先 【保護兒童/01】檢視性侵兒童法令 保護兒童的後盾抑或裝點門面的擺設?


ADVERTISEMENT
热门新闻





百格视频






ADVERTISEMENT





















